Recent from talks
Diffuse axonal injury
Knowledge base stats:
Talk channels stats:
Members stats:
Diffuse axonal injury
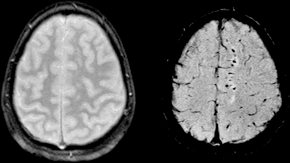
Diffuse axonal injury (DAI) is a brain injury in which scattered lesions occur over a widespread area in white matter tracts as well as grey matter. DAI is one of the most common and devastating types of traumatic brain injury and is a major cause of unconsciousness and persistent vegetative state after severe head trauma. It occurs in about half of all cases of severe head trauma and may be the primary damage that occurs in concussion. The outcome is frequently coma, with over 90% of patients with severe DAI never regaining consciousness. Those who awaken from the coma often remain significantly impaired.
DAI can occur across the spectrum of traumatic brain injury (TBI) severity, wherein the burden of injury increases from mild to severe. Concussion may be a milder type of diffuse axonal injury.
DAI is the result of traumatic shearing forces that occur when the head is rapidly accelerated or decelerated, as may occur in car accidents, falls, and assaults. Vehicle accidents are the most frequent cause of DAI; it can also occur as the result of child abuse such as in shaken baby syndrome.
Immediate disconnection of axons may be observed in severe brain injury, but the major damage of DAI is delayed secondary axon disconnections, slowly developed over an extended time course. Tracts of axons, which appear white due to myelination, are referred to as white matter. Lesions in both grey and white matter are found in postmortem brains in CT and MRI exams.
Besides mechanical breakage of the axonal cytoskeleton, DAI pathology also includes secondary physiological changes, such as interrupted axonal transport, progressive swellings known as axonal varicosities, and degeneration. Recent studies have linked these changes to twisting and misalignment of broken axon microtubules, as well as tau protein and amyloid precursor protein (APP) deposition.
Lesions typically are found in the white matter of brains injured by DAI; these lesions vary in size from about 1–15 mm and are distributed in a characteristic pattern. DAI most commonly affects white matter in areas including the brain stem, the corpus callosum, and the cerebral hemispheres.
The lobes of the brain most likely to be injured are the frontal and temporal lobes. Other common locations for DAI include the white matter in the cerebral cortex, the superior cerebral peduncles, basal ganglia, thalamus, and deep hemispheric nuclei.[clarification needed] These areas may be more easily damaged because of the difference in density between them and the other regions of the brain.
DAI is characterized by axonal separation, in which the axon is torn at the site of stretch and the part distal to the tear degrades by a process known as Wallerian degeneration. While it was once thought that the main cause of axonal separation was tearing due to mechanical forces during the trauma event, it is now understood that axons are not typically torn upon impact; rather, secondary biochemical cascades, which occur in response to the primary injury (which occurs as the result of mechanical forces at the moment of trauma) and take place hours to days after the initial injury, are largely responsible for the damage to axons.
Hub AI
Diffuse axonal injury AI simulator
(@Diffuse axonal injury_simulator)
Diffuse axonal injury
Diffuse axonal injury (DAI) is a brain injury in which scattered lesions occur over a widespread area in white matter tracts as well as grey matter. DAI is one of the most common and devastating types of traumatic brain injury and is a major cause of unconsciousness and persistent vegetative state after severe head trauma. It occurs in about half of all cases of severe head trauma and may be the primary damage that occurs in concussion. The outcome is frequently coma, with over 90% of patients with severe DAI never regaining consciousness. Those who awaken from the coma often remain significantly impaired.
DAI can occur across the spectrum of traumatic brain injury (TBI) severity, wherein the burden of injury increases from mild to severe. Concussion may be a milder type of diffuse axonal injury.
DAI is the result of traumatic shearing forces that occur when the head is rapidly accelerated or decelerated, as may occur in car accidents, falls, and assaults. Vehicle accidents are the most frequent cause of DAI; it can also occur as the result of child abuse such as in shaken baby syndrome.
Immediate disconnection of axons may be observed in severe brain injury, but the major damage of DAI is delayed secondary axon disconnections, slowly developed over an extended time course. Tracts of axons, which appear white due to myelination, are referred to as white matter. Lesions in both grey and white matter are found in postmortem brains in CT and MRI exams.
Besides mechanical breakage of the axonal cytoskeleton, DAI pathology also includes secondary physiological changes, such as interrupted axonal transport, progressive swellings known as axonal varicosities, and degeneration. Recent studies have linked these changes to twisting and misalignment of broken axon microtubules, as well as tau protein and amyloid precursor protein (APP) deposition.
Lesions typically are found in the white matter of brains injured by DAI; these lesions vary in size from about 1–15 mm and are distributed in a characteristic pattern. DAI most commonly affects white matter in areas including the brain stem, the corpus callosum, and the cerebral hemispheres.
The lobes of the brain most likely to be injured are the frontal and temporal lobes. Other common locations for DAI include the white matter in the cerebral cortex, the superior cerebral peduncles, basal ganglia, thalamus, and deep hemispheric nuclei.[clarification needed] These areas may be more easily damaged because of the difference in density between them and the other regions of the brain.
DAI is characterized by axonal separation, in which the axon is torn at the site of stretch and the part distal to the tear degrades by a process known as Wallerian degeneration. While it was once thought that the main cause of axonal separation was tearing due to mechanical forces during the trauma event, it is now understood that axons are not typically torn upon impact; rather, secondary biochemical cascades, which occur in response to the primary injury (which occurs as the result of mechanical forces at the moment of trauma) and take place hours to days after the initial injury, are largely responsible for the damage to axons.
Recent media