Recent from talks
Transmembrane protein
Knowledge base stats:
Talk channels stats:
Members stats:
Transmembrane protein
A transmembrane protein is a type of integral membrane protein that spans the entirety of the cell membrane. Many transmembrane proteins function as gateways to permit the transport of specific substances across the membrane. They frequently undergo significant conformational changes to move a substance through the membrane. They are usually highly hydrophobic and aggregate and precipitate in water. They require detergents or nonpolar solvents for extraction, although some of them (beta-barrels) can be also extracted using denaturing agents.
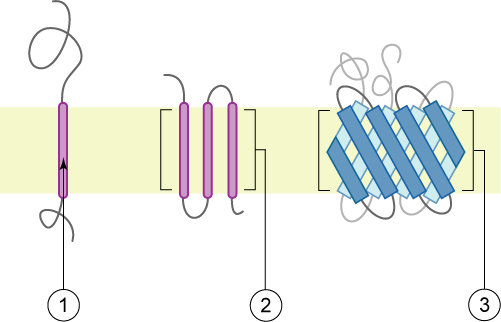
The peptide sequence that spans the membrane, or the transmembrane segment, is largely hydrophobic and can be visualized using the hydropathy plot. Depending on the number of transmembrane segments, transmembrane proteins can be classified as single-pass membrane proteins, or as multipass membrane proteins. Some other integral membrane proteins are called monotopic, meaning that they are also permanently attached to the membrane, but do not pass through it.
There are two basic types of transmembrane proteins: alpha-helical and beta barrels. Alpha-helical proteins are present in the inner membranes of bacterial cells or the plasma membrane of eukaryotic cells, and sometimes in the bacterial outer membrane. This is the major category of transmembrane proteins. In humans, 27% of all proteins have been estimated to be alpha-helical membrane proteins. Beta-barrel proteins are so far found only in outer membranes of gram-negative bacteria, cell walls of gram-positive bacteria, outer membranes of mitochondria and chloroplasts, or can be secreted as pore-forming toxins. All beta-barrel transmembrane proteins have simplest up-and-down topology, which may reflect their common evolutionary origin and similar folding mechanism.
In addition to the protein domains, there are unusual transmembrane elements formed by peptides. A typical example is gramicidin A, a peptide that forms a dimeric transmembrane β-helix. This peptide is secreted by gram-positive bacteria as an antibiotic. A transmembrane polyproline-II helix has not been reported in natural proteins. Nonetheless, this structure was experimentally observed in specifically designed artificial peptides.
This classification refers to the position of the protein N- and C-termini on the different sides of the lipid bilayer. Types I, II, III and IV are single-pass molecules. Type I transmembrane proteins are anchored to the lipid membrane with a stop-transfer anchor sequence and have their N-terminal domains targeted to the endoplasmic reticulum (ER) lumen during synthesis (and the extracellular space, if mature forms are located on cell membranes). Type II and III are anchored with a signal-anchor sequence, with type II being targeted to the ER lumen with its C-terminal domain, while type III have their N-terminal domains targeted to the ER lumen. Type IV is subdivided into IV-A, with their N-terminal domains targeted to the cytosol and IV-B, with an N-terminal domain targeted to the lumen. The implications for the division in the four types are especially manifest at the time of translocation and ER-bound translation, when the protein has to be passed through the ER membrane in a direction dependent on the type.[citation needed]
Membrane protein structures can be determined by X-ray crystallography, electron microscopy or NMR spectroscopy. The most common tertiary structures of these proteins are transmembrane helix bundle and beta barrel. The portion of the membrane proteins that are attached to the lipid bilayer (see annular lipid shell) consist mostly of hydrophobic amino acids.
Membrane proteins which have hydrophobic surfaces, are relatively flexible and are expressed at relatively low levels. This creates difficulties in obtaining enough protein and then growing crystals. Hence, despite the significant functional importance of membrane proteins, determining atomic resolution structures for these proteins is more difficult than globular proteins. As of January 2013 less than 0.1% of protein structures determined were membrane proteins despite being 20–30% of the total proteome. Due to this difficulty and the importance of this class of proteins methods of protein structure prediction based on hydropathy plots, the positive inside rule and other methods have been developed.
Transmembrane alpha-helical (α-helical) proteins are unusually stable judging from thermal denaturation studies, because they do not unfold completely within the membranes (the complete unfolding would require breaking down too many α-helical H-bonds in the nonpolar media). On the other hand, these proteins easily misfold, due to non-native aggregation in membranes, transition to the molten globule states, formation of non-native disulfide bonds, or unfolding of peripheral regions and nonregular loops that are locally less stable.
Hub AI
Transmembrane protein AI simulator
(@Transmembrane protein_simulator)
Transmembrane protein
A transmembrane protein is a type of integral membrane protein that spans the entirety of the cell membrane. Many transmembrane proteins function as gateways to permit the transport of specific substances across the membrane. They frequently undergo significant conformational changes to move a substance through the membrane. They are usually highly hydrophobic and aggregate and precipitate in water. They require detergents or nonpolar solvents for extraction, although some of them (beta-barrels) can be also extracted using denaturing agents.
The peptide sequence that spans the membrane, or the transmembrane segment, is largely hydrophobic and can be visualized using the hydropathy plot. Depending on the number of transmembrane segments, transmembrane proteins can be classified as single-pass membrane proteins, or as multipass membrane proteins. Some other integral membrane proteins are called monotopic, meaning that they are also permanently attached to the membrane, but do not pass through it.
There are two basic types of transmembrane proteins: alpha-helical and beta barrels. Alpha-helical proteins are present in the inner membranes of bacterial cells or the plasma membrane of eukaryotic cells, and sometimes in the bacterial outer membrane. This is the major category of transmembrane proteins. In humans, 27% of all proteins have been estimated to be alpha-helical membrane proteins. Beta-barrel proteins are so far found only in outer membranes of gram-negative bacteria, cell walls of gram-positive bacteria, outer membranes of mitochondria and chloroplasts, or can be secreted as pore-forming toxins. All beta-barrel transmembrane proteins have simplest up-and-down topology, which may reflect their common evolutionary origin and similar folding mechanism.
In addition to the protein domains, there are unusual transmembrane elements formed by peptides. A typical example is gramicidin A, a peptide that forms a dimeric transmembrane β-helix. This peptide is secreted by gram-positive bacteria as an antibiotic. A transmembrane polyproline-II helix has not been reported in natural proteins. Nonetheless, this structure was experimentally observed in specifically designed artificial peptides.
This classification refers to the position of the protein N- and C-termini on the different sides of the lipid bilayer. Types I, II, III and IV are single-pass molecules. Type I transmembrane proteins are anchored to the lipid membrane with a stop-transfer anchor sequence and have their N-terminal domains targeted to the endoplasmic reticulum (ER) lumen during synthesis (and the extracellular space, if mature forms are located on cell membranes). Type II and III are anchored with a signal-anchor sequence, with type II being targeted to the ER lumen with its C-terminal domain, while type III have their N-terminal domains targeted to the ER lumen. Type IV is subdivided into IV-A, with their N-terminal domains targeted to the cytosol and IV-B, with an N-terminal domain targeted to the lumen. The implications for the division in the four types are especially manifest at the time of translocation and ER-bound translation, when the protein has to be passed through the ER membrane in a direction dependent on the type.[citation needed]
Membrane protein structures can be determined by X-ray crystallography, electron microscopy or NMR spectroscopy. The most common tertiary structures of these proteins are transmembrane helix bundle and beta barrel. The portion of the membrane proteins that are attached to the lipid bilayer (see annular lipid shell) consist mostly of hydrophobic amino acids.
Membrane proteins which have hydrophobic surfaces, are relatively flexible and are expressed at relatively low levels. This creates difficulties in obtaining enough protein and then growing crystals. Hence, despite the significant functional importance of membrane proteins, determining atomic resolution structures for these proteins is more difficult than globular proteins. As of January 2013 less than 0.1% of protein structures determined were membrane proteins despite being 20–30% of the total proteome. Due to this difficulty and the importance of this class of proteins methods of protein structure prediction based on hydropathy plots, the positive inside rule and other methods have been developed.
Transmembrane alpha-helical (α-helical) proteins are unusually stable judging from thermal denaturation studies, because they do not unfold completely within the membranes (the complete unfolding would require breaking down too many α-helical H-bonds in the nonpolar media). On the other hand, these proteins easily misfold, due to non-native aggregation in membranes, transition to the molten globule states, formation of non-native disulfide bonds, or unfolding of peripheral regions and nonregular loops that are locally less stable.
Recent media