

Community hub
Recent from talks
Contribute something
Nothing was collected or created yet.
Tacrolimus
View on Wikipedia
 | |
 | |
| Clinical data | |
|---|---|
| Trade names | Prograf, Advagraf, Protopic, Envarsus, others |
| Other names | FK-506, fujimycin |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a601117 |
| License data |
|
| Pregnancy category |
|
| Routes of administration | Topical, by mouth, intravenous |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 24% (5–67%), less after eating food rich in fat |
| Protein binding | ≥98.8% |
| Metabolism | Liver CYP3A4, CYP3A5 |
| Elimination half-life | 11.3 h for transplant patients (range 3.5–40.6 h) |
| Excretion | Mostly fecal |
| Identifiers | |
| |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| PDB ligand | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.155.367 |
| Chemical and physical data | |
| Formula | C44H69NO12 |
| Molar mass | 804.031 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
| | |
Tacrolimus, sold under the brand name Prograf among others, is an immunosuppressive drug. After an allogenic organ transplant, the risk of organ rejection is moderate; tacrolimus is used to lower the risk of organ rejection. Tacrolimus is also sold as a topical medication for treating T cell-mediated diseases, such as eczema and psoriasis. For example, it is prescribed for severe refractory uveitis after a bone marrow transplant, exacerbations of minimal change disease, Kimura's disease, and vitiligo. It can be used to treat dry eye syndrome in cats and dogs.[6][7]
Tacrolimus inhibits calcineurin, which is involved in the production of interleukin-2, a molecule that promotes the development and proliferation of T cells, as part of the body's learned (or adaptive) immune response.
Chemically, it is a macrolide lactone[8] that was first discovered in 1987, from the fermentation broth of a Japanese soil sample that contained the bacterium Streptomyces tsukubensis. It is on the World Health Organization's List of Essential Medicines.[9] In 2021, it was the 296th most commonly prescribed medication in the United States, with more than 500,000 prescriptions.[10][11]
Medical uses
[edit]Organ transplantation
[edit]It has similar immunosuppressive properties to ciclosporin, but is much more potent. Immunosuppression with tacrolimus was associated with a significantly lower rate of acute rejection compared with ciclosporin-based immunosuppression (30.7% vs 46.4%) in one study.[12] Clinical outcome is better with tacrolimus than with ciclosporin during the first year of liver transplantation.[13][14] Long-term outcome has not been improved to the same extent. Tacrolimus is normally prescribed as part of a post-transplant cocktail including steroids, mycophenolate, and IL-2 receptor inhibitors such as basiliximab. Dosages are titrated to target blood levels at specific times after medication administration.[15]
Skin
[edit]
As an ointment, tacrolimus is used in the treatment of dermatitis (eczema), in particular atopic dermatitis, if topical corticosteroids and moisturisers fail in helping.[16][17] It suppresses inflammation in a similar way to steroids, and is equally as effective as a mid-potency steroid. An important advantage of tacrolimus is that, unlike steroids, it does not cause skin thinning (atrophy), or other steroid related side effects.[18][17]
It is applied on the active lesions until they heal off, but may also be used continuously in low doses (twice a week), and applied to the thinner skin over the face and eyelids.[citation needed] Clinical trials of up to one year have been conducted. Recently it has also been used to treat segmental vitiligo in children, especially in areas on the face.[19]
Eyes
[edit]Tacrolimus solution, as drops, is sometimes prescribed by veterinarians for keratoconjunctivitis, and other dry eye maladies, in the eyes of domestic cats, dogs, and horses.[20] It has been studied for use in human eyes.[21][22]
Contraindications and precautions
[edit]Contraindications and precautions include:[23]
- Hepatic disease
- Immunosuppression
- Infants
- Infection
- Neoplastic disease, such as:
- Oliguria
- Pregnancy
- QT interval prolongation
- Sunlight (UV) exposure
- Grapefruit juice[24]
Topical use
[edit]- Occlusive dressing
- Known or suspected malignant lesions
- Netherton's syndrome or similar skin diseases
- Certain skin infections[18]
Side effects
[edit]By mouth or intravenous use
[edit]Side effects can be severe and include infection, cardiac damage, hypertension, blurred vision, liver and kidney problems (tacrolimus nephrotoxicity),[25] hyperkalemia, hypomagnesemia, hyperglycemia, diabetes mellitus, itching, lung damage (sirolimus also causes lung damage),[26] and various neuropsychiatric problems such as loss of appetite, insomnia, posterior reversible encephalopathy syndrome, confusion, weakness, depression, vivid nightmares, cramps, neuropathy, seizures, tremors, and catatonia.[27]
In addition, it may potentially increase the severity of existing fungal or infectious conditions such as herpes zoster or polyoma viral infections.[23]
Carcinogenesis and mutagenesis
[edit]In people receiving immunosuppressants to reduce transplant graft rejection, an increased risk of malignancy (cancer) is a recognised complication.[23] The most common cancers are non-Hodgkin's lymphoma[28] and skin cancers. The risk appears to be related to the intensity and duration of treatment.
Topical use
[edit]The most common adverse events associated with the use of topical tacrolimus ointments, especially if used over a wide area, include a burning or itching sensation on the initial applications, with increased sensitivity to sunlight and heat on the affected areas.[citation needed] Less common are flu-like symptoms, headache, cough, and burning eyes.[29]
Cancer risks
[edit]Tacrolimus and a related drug for eczema (pimecrolimus) were suspected of carrying a cancer risk, though the matter is still a subject of controversy. The FDA issued a health warning in March 2005 for the drug, based on animal models and a small number of patients. Until further human studies yield more conclusive results, the FDA recommends that users be advised of the potential risks. However, current practice by UK dermatologists is not to consider this a significant real concern and they are increasingly recommending the use of these new drugs.[30] A 2023 systematic review and meta-analysis published in The Lancet Child & Adolescent Health concluded with moderate-certainty evidence that the two drugs were not associated with any increased risk of cancer.[31]
In November 2024, International Agency for Research on Cancer (IARC) classified hydrochlorothiazide, voriconazole and tacrolimus as group 1 carcinogens.[32][33]
Interactions
[edit]Also like cyclosporin, it has a wide range of interactions. Tacrolimus is primarily metabolised by the cytochrome P450 system of liver enzymes, and there are many substances that interact with this system and induce or inhibit the system's metabolic activity.[23]
Interactions include that with grapefruit which increases tacrolimus plasma concentrations. As infections are a major cause of morbidity and mortality in the post-transplant patient, the most commonly[citation needed] reported interactions include interactions with anti-microbial drugs. Macrolide antibiotics including erythromycin and clarithromycin, as well as several of the newer classes of antifungals, especially of the azole class (fluconazole, voriconazole), increase tacrolimus levels by competing for cytochrome enzymes.[23]
Pharmacology
[edit]Mechanism of action
[edit]
Tacrolimus is a macrolide calcineurin inhibitor. In T cells, activation of the T cell receptor normally increases intracellular calcium, which acts via calmodulin to activate calcineurin. Calcineurin then dephosphorylates the transcription factor nuclear factor of activated T cells (NF-AT), which moves to the nucleus of the T cell and increases the activity of genes coding for IL-2 and related cytokines. Tacrolimus prevents the dephosphorylation of NF-AT.[34]
In detail, tacrolimus reduces peptidylprolyl isomerase activity by binding to the immunophilin FKBP12 (FK506 binding protein), creating a new complex. This FKBP12–FK506 complex interacts with and inhibits calcineurin, thus inhibiting both T lymphocyte signal transduction and IL-2 transcription.[35] Although this activity is similar to that of cyclosporin, the incidence of acute rejection is reduced by tacrolimus use over cyclosporin use.[12] Although short-term immunosuppression concerning patient and graft survival is found to be similar between the two drugs, tacrolimus results in a more favorable lipid profile, and this may have important long-term implications given the prognostic influence of rejection on graft survival.[36]
Pharmacokinetics
[edit]Oral tacrolimus is slowly absorbed in the gastrointestinal tract, with a total bioavailability of 20 to 25% (but with variations from 5 to 67%) and highest blood plasma concentrations (Cmax) reached after one to three hours. Taking the drug together with a meal, especially one rich in fat, slows down resorption and reduces bioavailability. In the blood, tacrolimus is mainly bound to erythrocytes; only 5% are found in the plasma, of which more than 98.8% are bound to plasma proteins.[23][37]
The substance is metabolized in the liver, mainly via CYP3A, and in the intestinal wall. All metabolites found in the circulation are inactive. Biological half-life varies widely and seems to be higher for healthy persons (43 hours on average) than for patients with liver transplants (12 hours) or kidney transplants (16 hours), due to differences in clearance. Tacrolimus is predominantly eliminated via the faeces in form of its metabolites.[23][37]
When applied locally on eczema, tacrolimus has little to no bioavailability.[23]
Pharmacogenetics
[edit]The predominant enzyme responsible for metabolism of tacrolimus is CYP3A5. Genetic variations within CYP3A5 that result in changes to the activity of the CYP3A5 protein can affect concentrations of tacrolimus within the body. In particular, individuals who are homozygous for the G allele at the single nucleotide polymorphism (SNP) rs776746 (also known as CYP3A5 *3/*3) have a non-functional CYP3A5 protein. The frequency of the G allele varies worldwide, from 4% in some African populations to 80–90% in Caucasian populations.[38] Across a large number of studies, individuals homozygous for the G allele have been shown to have higher concentrations of tacrolimus and require lower doses of the drug, as compared to individuals who are not homozygous for the G allele. Achieving target concentrations of tacrolimus is important – if levels are too low, then there is a risk of transplant rejection, if levels are too high, there is a risk of drug toxicities. There is evidence to suggest that dosing patients based on rs776746 genotype can result in faster and more frequent achievement of target tacrolimus levels. However, there is a lack of consistent evidence as to whether dosing based on rs776746 genotype results in improved clinical outcomes (such as a decreased risk for transplant rejection or drug toxicities), likely because patients taking tacrolimus are subject to therapeutic drug monitoring.[39][40][41][42]
Studies have shown that genetic polymorphisms of genes other than CYP3A5, such as NR1I2[43][44] (encoding PXR), also significantly influence the pharmacokinetics of tacrolimus.
History
[edit]Tacrolimus was discovered in 1987 by a Japanese team led by pharmacologist Tohru Kino; it was among the first macrolide immunosuppressants discovered, preceded by the discovery of rapamycin (sirolimus) on Rapa Nui (Easter Island) in 1975.[45] It is produced by a soil bacterium, Streptomyces tsukubensis.[46] The name tacrolimus is derived from "Tsukuba macrolide immunosuppressant".[47]
The early development (investigational new drug phase) of tacrolimus, called at the time by the development code FK-506, happened in the next several years. A firsthand account of that process is given in Thomas Starzl's 1992 memoir.[48]
Tacrolimus was first approved by the US Food and Drug Administration (FDA) in 1994,[49][50] for use in liver transplantation; the indications were extended to include kidney transplants.[51] The first generic version of tacrolimus (capsule for oral route) was approved in the US in 2009.[52] A generic version of tacrolimus for injection was approved in the US in 2017.[53]
Tacrolimus was approved for medical use in the European Union in 2002, for the treatment of moderate to severe atopic dermatitis.[54] In 2007, the indications were expanded to include the prophylaxis of transplant rejection in adult kidney or liver allograft recipients and the treatment of allograft rejection resistant to treatment with other immunosuppressive medicinal products in adults.[55] In 2009, the indications were expanded to include the prophylaxis of transplant rejection in adult and paediatric, kidney, liver or heart allograft recipients and the treatment of allograft rejection resistant to treatment with other immunosuppressive medicinal products in adults and children.[56]
Available forms
[edit]A branded version of the drug is owned by Astellas Pharma, and is sold under the brand name Prograf, given twice daily. A number of other manufacturers hold marketing authorisation for alternative brands of the twice-daily formulation.[57]
Once-daily formulations with marketing authorisation include Advagraf (Astellas Pharma) and Envarsus (marketed as Envarsus XR in US by Veloxis Pharmaceuticals and marketed in Europe by Chiesi).[57] These formulations are intended to reduce pharmacokinetic variation in blood levels and facilitate compliance with dosing.[citation needed]
The topical formulation is marketed by LEO Pharma under the name Protopic.[57]
Biosynthesis
[edit]
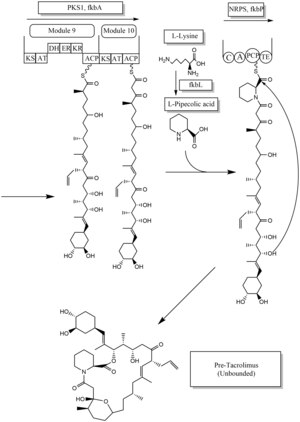

The biosynthesis of tacrolimus is hybrid synthesis of both type 1 polyketide synthases (PKS 1) and nonribosomal peptide syntheses (NRPS). The research shows the hybrid synthesis consists of ten modules of type 1 polyketide synthase and one module of nonribosomal peptide synthase. The synthetic enzymes for tacrolimus are found in 19 gene clusters named fkb. The 19 genes are fkbQ, fkbN, fkbM, fkbD, fkbA, fkbP, fkbO, fkbB, fkbC, fkbL, fkbK, fkbJ, fkbI, fkbH, fkbG, allD, allR, allK and allA.[58]
There are several possible ways of biosynthesis of tacrolimus. The fundamental units for biosynthesis are following: one molecule of 4,5-dihydroxycyclohex-1-enecarboxylic acid (DHCHC) as a starter unit, four molecules of malonyl-CoA, five molecules of methylmalonyl-CoA, one molecule of allylmalonyl-CoA as elongation units. However, two molecules of malonyl-CoA are able to be replaced by two molecules of methoxymalonyl CoA. Once two malonyl-CoA molecules are replaced, post-synthase tailoring steps are no longer required where two methoxymalonyl CoA molecules are substituted. The biosynthesis of methoxymalonyl CoA to Acyl Carrier protein is proceeded by five enzymes (fkbG, fkbH, fkbI, fkbJ, and fkbK). Allylmalonyl-CoA is also able to be replaced by propionylmalonyl-CoA.[58]
The starter unit, DHCHC from the chorismic acid is formed by fkbO enzyme and loaded onto CoA-ligase domain (CoL). Then, it proceeds to NADPH dependent reduction(ER). Three enzymes, fkbA,B,C enforce processes from the loading module to the module 10, the last step of PKS 1. fkbB enzyme is responsible of allylmalonyl-CoA synthesis or possibly propionylmalonyl-CoA at C21, which it is an unusual step of general PKS 1. As mentioned, if two methoxymalonyl CoA molecules are substituted for two malonyl-CoA molecules, they will take place in module 7 and 8 (C13 and C15), and fkbA enzyme will enforce this process. After the last step (module 10) of PKS 1, one molecule of L-pipecolic acid formed from L-lysine and catalyzed through fkbL enzyme synthesizes with the molecule from the module 10. The process of L-pipecolic acid synthesis is NRPS enforced by fkbP enzyme. After synthesizing the entire subunits, the molecule is cyclized. After the cyclization, the pre-tacrolimus molecule goes through the post-synthase tailoring steps such as oxidation and S-adenosyl methionine. Particularly fkbM enzyme is responsible of alcohol methylation targeting the alcohol of DHCHC starter unit (Carbon number 31 depicted in brown), and fkbD enzyme is responsible of C9 (depicted in green). After these tailoring steps, the tacrolimus molecule becomes biologically active.[58][59][60]
Research
[edit]Lupus nephritis
[edit]Tacrolimus has been shown to reduce the risk of serious infections while also increasing remission of kidney function in lupus nephritis.[61][62]
Ulcerative colitis
[edit]Tacrolimus has been used to suppress the inflammation associated with ulcerative colitis (UC), a form of inflammatory bowel disease. Although almost exclusively used in trial cases only, tacrolimus has shown to be significantly effective in the suppression of flares of UC.[63] A 2022 updated Cochrane systematic review found that tacrolimus may be superior to placebo in achieving remission and improvement in UC.[64]
References
[edit]- ^ "Tacrolimus Use During Pregnancy". Drugs.com. 3 October 2019. Retrieved 29 April 2020.
- ^ "Drug and medical device highlights 2019: Helping you maintain and improve your health". Health Canada. 18 November 2020. Retrieved 28 March 2024.
- ^ "Prograf- tacrolimus capsule, gelatin coated Prograf- tacrolimus injection, solution Prograf- tacrolimus granule, for suspension". DailyMed. Retrieved 16 July 2021.
- ^ "Advagraf EPAR". European Medicines Agency. 17 September 2018. Retrieved 16 July 2021.
- ^ "Protopic EPAR". European Medicines Agency. 17 September 2018. Retrieved 16 July 2021.
- ^ Berdoulay A, English RV, Nadelstein B (2005). "Effect of topical 0.02% tacrolimus aqueous suspension on tear production in dogs with keratoconjunctivitis sicca". Veterinary Ophthalmology. 8 (4): 225–232. doi:10.1111/j.1463-5224.2005.00390.x. PMID 16008701.
- ^ "Tacrolimus for Dogs and Cats".
- ^ Baldo A, Cafiero M, Di Caterino P, Di Costanzo L (January 2009). "Tacrolimus ointment in the management of atopic dermatitis". Clinical, Cosmetic and Investigational Dermatology. 2: 1–7. doi:10.2147/ccid.s3378. PMC 3047924. PMID 21436963.
- ^ World Health Organization (2023). The selection and use of essential medicines 2023: web annex A: World Health Organization model list of essential medicines: 23rd list (2023). Geneva: World Health Organization. hdl:10665/371090. WHO/MHP/HPS/EML/2023.02.
- ^ "The Top 300 of 2021". ClinCalc. Archived from the original on 15 January 2024. Retrieved 14 January 2024.
- ^ "Tacrolimus - Drug Usage Statistics". ClinCalc. Retrieved 14 January 2024.
- ^ a b McCauley J (19 May 2004). "Long-Term Graft Survival In Kidney Transplant Recipients". Slide Set Series on Analyses of Immunosuppressive Therapies. Medscape. Retrieved 6 June 2006.
- ^ Haddad EM, McAlister VC, Renouf E, Malthaner R, Kjaer MS, Gluud LL (October 2006). McAlister V (ed.). "Cyclosporin versus tacrolimus for liver transplanted patients". The Cochrane Database of Systematic Reviews. 2006 (4) CD005161. doi:10.1002/14651858.CD005161.pub2. PMC 8865611. PMID 17054241.
- ^ O'Grady JG, Burroughs A, Hardy P, Elbourne D, Truesdale A (October 2002). "Tacrolimus versus microemulsified ciclosporin in liver transplantation: the TMC randomised controlled trial". Lancet. 360 (9340): 1119–1125. doi:10.1016/S0140-6736(02)11196-2. PMID 12387959. S2CID 10417106.
- ^ Lee MN, Butani L (June 2007). "Improved pharmacokinetic monitoring of tacrolimus exposure after pediatric renal transplantation". Pediatric Transplantation. 11 (4): 388–393. doi:10.1111/j.1399-3046.2006.00618.x. PMID 17493218. S2CID 23530214.
- ^ Cury Martins J, Martins C, Aoki V, Gois AF, Ishii HA, da Silva EM (July 2015). "Topical tacrolimus for atopic dermatitis". The Cochrane Database of Systematic Reviews. 2015 (7) CD009864. doi:10.1002/14651858.CD009864.pub2. PMC 6461158. PMID 26132597.
- ^ a b Devasenapathy N, Chu A, Wong M, Srivastava A, Ceccacci R, Lin C, et al. (January 2023). "Cancer risk with topical calcineurin inhibitors, pimecrolimus and tacrolimus, for atopic dermatitis: a systematic review and meta-analysis". The Lancet. Child & Adolescent Health. 7 (1): 13–25. doi:10.1016/S2352-4642(22)00283-8. PMID 36370744. S2CID 253470127.
- ^ a b Haberfeld, H, ed. (2015). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag. Protopic.
- ^ Silverberg NB, Lin P, Travis L, Farley-Li J, Mancini AJ, Wagner AM, et al. (November 2004). "Tacrolimus ointment promotes repigmentation of vitiligo in children: a review of 57 cases". Journal of the American Academy of Dermatology. 51 (5): 760–766. doi:10.1016/j.jaad.2004.05.036. PMID 15523355.
- ^ "Tacrolimus, Ophthalmic" (PDF). Plumbs Veterinary Medication Guides. 2017. Archived from the original (PDF) on 18 December 2022. Retrieved 5 August 2022.
- ^ Naves FE, Sakassegawa E (10 May 2013). "Treatment of Dry Eye Using 0.03% Tacrolimus Eye Drops". U.S. National Library of Medicine. Retrieved 5 August 2022.
- ^ Yazu H, Fukagawa K, Shimizu E, Sato Y, Fujishima H (February 2021). "Long-term outcomes of 0.1% tacrolimus eye drops in eyes with severe allergic conjunctival diseases". Allergy, Asthma, and Clinical Immunology. 17 (1) 11. doi:10.1186/s13223-021-00513-w. PMC 7852099. PMID 33522964.
- ^ a b c d e f g h Haberfeld, H, ed. (2015). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag. Prograf.
- ^ Fukatsu S, Fukudo M, Masuda S, Yano I, Katsura T, Ogura Y, et al. (April 2006). "Delayed effect of grapefruit juice on pharmacokinetics and pharmacodynamics of tacrolimus in a living-donor liver transplant recipient". Drug Metabolism and Pharmacokinetics. 21 (2): 122–125. doi:10.2133/dmpk.21.122. PMID 16702731.
- ^ Naesens M, Kuypers DR, Sarwal M (February 2009). "Calcineurin inhibitor nephrotoxicity". Clinical Journal of the American Society of Nephrology. 4 (2): 481–508. doi:10.2215/CJN.04800908. PMID 19218475.
- ^ Miwa Y, Isozaki T, Wakabayashi K, Odai T, Matsunawa M, Yajima N, et al. (2008). "Tacrolimus-induced lung injury in a rheumatoid arthritis patient with interstitial pneumonitis". Modern Rheumatology. 18 (2): 208–211. doi:10.1007/s10165-008-0034-3. PMID 18306979. S2CID 39537409.
- ^ O'Donnell MM, Williams JP, Weinrieb R, Denysenko L (2007). "Catatonic mutism after liver transplant rapidly reversed with lorazepam". General Hospital Psychiatry. 29 (3): 280–281. doi:10.1016/j.genhosppsych.2007.01.004. PMID 17484951.
- ^ "Key Statistics for Non-Hodgkin Lymphoma". www.cancer.org. Retrieved 19 February 2020.
- ^ Hanifin JM, Paller AS, Eichenfield L, Clark RA, Korman N, Weinstein G, et al. (August 2005). "Efficacy and safety of tacrolimus ointment treatment for up to 4 years in patients with atopic dermatitis". Journal of the American Academy of Dermatology. 53 (2 Suppl 2): S186 – S194. doi:10.1016/j.jaad.2005.04.062. PMID 16021174.
- ^ Cox NH, Smith CH (December 2002). "Advice to dermatologists re topical tacrolimus" (PDF). Therapy Guidelines Committee. British Association of Dermatologists. Archived from the original (PDF) on 13 December 2013.
- ^ Devasenapathy N, Chu A, Wong M, Srivastava A, Ceccacci R, Lin C, et al. (January 2023). "Cancer risk with topical calcineurin inhibitors, pimecrolimus and tacrolimus, for atopic dermatitis: a systematic review and meta-analysis". The Lancet. Child & Adolescent Health. 7 (1): 13–25. doi:10.1016/S2352-4642(22)00283-8. PMID 36370744. S2CID 253470127.
- ^ Cogliano VJ, Corsini E, Fournier A, Nelson HH, Sergi CM, Antunes AMM, et al. (29 November 2024). "Carcinogenicity of hydrochlorothiazide, voriconazole, and tacrolimus". The Lancet Oncology. 26 (1): 15–16. doi:10.1016/S1470-2045(24)00685-5. PMC 12174844. PMID 39622256.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ^ "List of Classifications". IARC. Retrieved 12 April 2025.
- ^ Ganong WF (8 March 2005). Review of medical physiology (22nd ed.). Lange medical books. p. 530. ISBN 978-0-07-144040-0.
- ^ Liu J, Farmer JD, Lane WS, Friedman J, Weissman I, Schreiber SL (August 1991). "Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes". Cell. 66 (4): 807–815. doi:10.1016/0092-8674(91)90124-H. PMID 1715244. S2CID 22094672.
- ^ Abou-Jaoude MM, Najm R, Shaheen J, Nawfal N, Abboud S, Alhabash M, et al. (September 2005). "Tacrolimus (FK506) versus cyclosporine microemulsion (neoral) as maintenance immunosuppression therapy in kidney transplant recipients". Transplantation Proceedings. 37 (7): 3025–3028. doi:10.1016/j.transproceed.2005.08.040. PMID 16213293.
- ^ a b Dinnendahl, V, Fricke, U, eds. (2003). Arzneistoff-Profile (in German). Vol. 9 (18 ed.). Eschborn, Germany: Govi Pharmazeutischer Verlag. ISBN 978-3-7741-9846-3.
- ^ Bains RK. "Molecular diversity and population structure at the CYP3A5 gene in Africa" (PDF). University College London. Retrieved 13 June 2016.
- ^ Staatz CE, Tett SE (2004). "Clinical pharmacokinetics and pharmacodynamics of tacrolimus in solid organ transplantation". Clinical Pharmacokinetics. 43 (10): 623–653. doi:10.2165/00003088-200443100-00001. PMID 15244495. S2CID 33877550.
- ^ Staatz CE, Goodman LK, Tett SE (March 2010). "Effect of CYP3A and ABCB1 single nucleotide polymorphisms on the pharmacokinetics and pharmacodynamics of calcineurin inhibitors: Part I". Clinical Pharmacokinetics. 49 (3): 141–175. doi:10.2165/11317350-000000000-00000. PMID 20170205. S2CID 28346861.
- ^ Staatz CE, Goodman LK, Tett SE (April 2010). "Effect of CYP3A and ABCB1 single nucleotide polymorphisms on the pharmacokinetics and pharmacodynamics of calcineurin inhibitors: Part II". Clinical Pharmacokinetics. 49 (4): 207–221. doi:10.2165/11317550-000000000-00000. PMID 20214406. S2CID 27047235.
- ^ Barbarino JM, Staatz CE, Venkataramanan R, Klein TE, Altman RB (October 2013). "PharmGKB summary: cyclosporine and tacrolimus pathways". Pharmacogenetics and Genomics. 23 (10): 563–585. doi:10.1097/fpc.0b013e328364db84. PMC 4119065. PMID 23922006.
- ^ Benkali K, Prémaud A, Picard N, Rérolle JP, Toupance O, Hoizey G, et al. (1 January 2009). "Tacrolimus population pharmacokinetic-pharmacogenetic analysis and Bayesian estimation in renal transplant recipients". Clinical Pharmacokinetics. 48 (12): 805–816. doi:10.2165/11318080-000000000-00000. PMID 19902988. S2CID 19900291.
- ^ Choi Y, Jiang F, An H, Park HJ, Choi JH, Lee H (January 2017). "A pharmacogenomic study on the pharmacokinetics of tacrolimus in healthy subjects using the DMETTM Plus platform". The Pharmacogenomics Journal. 17 (1): 105–106. doi:10.1038/tpj.2016.85. PMID 27958377.
- ^ Kino T, Hatanaka H, Hashimoto M, Nishiyama M, Goto T, Okuhara M, et al. (September 1987). "FK-506, a novel immunosuppressant isolated from a Streptomyces. I. Fermentation, isolation, and physico-chemical and biological characteristics". The Journal of Antibiotics. 40 (9): 1249–1255. doi:10.7164/antibiotics.40.1249. PMID 2445721.
- ^ Pritchard DI (May 2005). "Sourcing a chemical succession for cyclosporin from parasites and human pathogens". Drug Discovery Today. 10 (10): 688–691. doi:10.1016/S1359-6446(05)03395-7. PMID 15896681. Supports source organism, but not team information
- ^ Ponner, B, Cvach, B (Fujisawa Pharmaceutical Co.): Protopic Update 2005
- ^ Starzl TE (1992). "Chapter 25: The drug with no name". The Puzzle People: Memoirs Of A Transplant Surgeon. University of Pittsburgh Press. pp. 288–308. doi:10.2307/j.ctt9qh63b. ISBN 978-0-8229-3714-2.
- ^ "Prograf: FDA-Approved Drugs". U.S. Food and Drug Administration (FDA). Archived from the original on 30 April 2017. Retrieved 29 April 2020.
- ^ "Prograf: FDA-Approved Drugs". U.S. Food and Drug Administration (FDA). Archived from the original on 30 April 2017. Retrieved 29 April 2020.
- ^ Tacrolimus (Systemic) Monograph. Accessed 16 December 2021.
- ^ "Drugs@FDA: FDA-Approved Drugs". www.accessdata.fda.gov. Retrieved 17 September 2025.
- ^ "Tacrolimus: FDA-Approved Drugs". U.S. Food and Drug Administration (FDA). Archived from the original on 8 April 2019. Retrieved 29 April 2020.
- ^ "Protopic EPAR". European Medicines Agency (EMA). 17 September 2018. Retrieved 29 April 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain.
- ^ "Advagraf EPAR". European Medicines Agency (EMA). 17 September 2018. Retrieved 29 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ "Modigraf EPAR". European Medicines Agency (EMA). 17 September 2018. Retrieved 29 April 2020.
- ^ a b c Joint Formulary Committee. "British National Formulary (online)". London: BMJ Group and Pharmaceutical Press. Retrieved 24 September 2015.
- ^ a b c Ordóñez-Robles M, Santos-Beneit F, Martín JF (May 2018). "Unraveling Nutritional Regulation of Tacrolimus Biosynthesis in Streptomyces tsukubaensis through omic Approaches". Antibiotics. 7 (2): 39. doi:10.3390/antibiotics7020039. PMC 6022917. PMID 29724001.
- ^ Chen D, Zhang L, Pang B, Chen J, Xu Z, Abe I, et al. (May 2013). "FK506 maturation involves a cytochrome p450 protein-catalyzed four-electron C-9 oxidation in parallel with a C-31 O-methylation". Journal of Bacteriology. 195 (9): 1931–1939. doi:10.1128/JB.00033-13. PMC 3624582. PMID 23435975.
- ^ Mo S, Ban YH, Park JW, Yoo YJ, Yoon YJ (December 2009). "Enhanced FK506 production in Streptomyces clavuligerus CKD1119 by engineering the supply of methylmalonyl-CoA precursor". Journal of Industrial Microbiology & Biotechnology. 36 (12): 1473–1482. doi:10.1007/s10295-009-0635-7. PMID 19756799. S2CID 32967249.
- ^ Singh JA, Hossain A, Kotb A, Wells G (September 2016). "Risk of serious infections with immunosuppressive drugs and glucocorticoids for lupus nephritis: a systematic review and network meta-analysis". BMC Medicine. 14 (1) 137. doi:10.1186/s12916-016-0673-8. PMC 5022202. PMID 27623861.
- ^ Singh JA, Hossain A, Kotb A, Wells GA (September 2016). "Comparative effectiveness of immunosuppressive drugs and corticosteroids for lupus nephritis: a systematic review and network meta-analysis". Systematic Reviews. 5 (1) 155. doi:10.1186/s13643-016-0328-z. PMC 5020478. PMID 27619512.
- ^ Baumgart DC, Pintoffl JP, Sturm A, Wiedenmann B, Dignass AU (May 2006). "Tacrolimus is safe and effective in patients with severe steroid-refractory or steroid-dependent inflammatory bowel disease--a long-term follow-up". The American Journal of Gastroenterology. 101 (5): 1048–1056. doi:10.1111/j.1572-0241.2006.00524.x. PMID 16573777. S2CID 10233231.
- ^ Gordon M, Sinopoulou V, Akobeng AK, Pana M, Gasiea R, Moran GW (April 2022). "Tacrolimus (FK506) for induction of remission in corticosteroid-refractory ulcerative colitis". The Cochrane Database of Systematic Reviews. 2022 (4) CD007216. doi:10.1002/14651858.CD007216.pub2. PMC 8987360. PMID 35388476.
Further reading
[edit]- Lv X, Qi J, Zhou M, Shi B, Cai C, Tang Y, et al. (June 2020). "Comparative efficacy of 20 graft-versus-host disease prophylaxis therapies for patients after hematopoietic stem-cell transplantation: A multiple-treatments network meta-analysis". Critical Reviews in Oncology/Hematology. 150 102944. doi:10.1016/j.critrevonc.2020.102944. PMID 32247246. S2CID 214794350.
External links
[edit]- "Tacrolimus Injection". MedlinePlus.
- "Tacrolimus Topical". MedlinePlus.
- Tacrolimus at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- "FDA Approves New Use of Transplant Drug Based on Real-World Evidence". U.S. Food and Drug Administration (FDA). 30 September 2021. Archived from the original on 16 July 2021.