

Community hub
Recent from talks
Contribute something
Nothing was collected or created yet.
Electrolysis
View on Wikipedia

| Electromagnetism |
|---|
 |
In chemistry and manufacturing, electrolysis is a technique that uses direct electric current (DC) to drive an otherwise non-spontaneous chemical reaction. Electrolysis is commercially important as a stage in the separation of elements from naturally occurring sources such as ores using an electrolytic cell. The voltage that is needed for electrolysis to occur is called the decomposition potential. The word "lysis" means to separate or break, so in terms, electrolysis would mean "breakdown via electricity."
Etymology
[edit]The word "electrolysis" was introduced by Michael Faraday in 1834,[1] using the Greek words ἤλεκτρον [ɛ̌ːlektron] "amber", which since the 17th century was associated with electrical phenomena, and λύσις [lýsis] meaning "dissolution". Nevertheless, electrolysis, as a tool to study chemical reactions and obtain pure elements, precedes the coinage of the term and formal description by Faraday.
History
[edit]In the early nineteenth century, William Nicholson and Anthony Carlisle sought to further Volta's experiments. They attached two wires to either side of a voltaic pile and placed the other ends in a tube filled with water. They noticed when the wires were brought together that each wire produced bubbles. One type was hydrogen, the other was oxygen.[2]
In 1785 a Dutch scientist named Martin van Marum created an electrostatic generator that he used to reduce tin, zinc and antimony from their salts using a process later known as electrolysis. Though he unknowingly produced electrolysis, it was not until 1800 when William Nicholson and Anthony Carlisle discovered how electrolysis works.[3]
In 1791 Luigi Galvani experimented with frog legs. He claimed that placing animal muscle between two dissimilar metal sheets resulted in electricity. Responding to these claims, Alessandro Volta conducted his own tests.[4][5] This would give insight to Humphry Davy's ideas on electrolysis. During preliminary experiments, Humphry Davy hypothesized that when two elements combine to form a compound, electrical energy is released. Humphry Davy would go on to create Decomposition Tables from his preliminary experiments on Electrolysis. The Decomposition Tables would give insight on the energies needed to break apart certain compounds.[6]
In 1817 Johan August Arfwedson determined there was another element, lithium, in some of his samples; however, he could not isolate the component. It was not until 1821 that William Thomas Brande used electrolysis to single it out. Two years later, he streamlined the process using lithium chloride and potassium chloride with electrolysis to produce lithium and lithium hydroxide.[7][8]
During the later years of Humphry Davy's research, Michael Faraday became his assistant. While studying the process of electrolysis under Humphry Davy, Michael Faraday discovered two laws of electrolysis.[5]
During the time of Maxwell and Faraday, concerns came about[vague] for electropositive and electronegative activities.[9]
In November 1875, Paul Émile Lecoq de Boisbaudran discovered gallium using electrolysis of gallium hydroxide, producing 3.4 mg of gallium. The following December, he presented his discovery of gallium to the Académie des sciences in Paris.[10]
On June 26, 1886, Ferdinand Frederick Henri Moissan finally felt comfortable performing electrolysis on anhydrous hydrogen fluoride to create a gaseous fluorine pure element. Before he used hydrogen fluoride, Henri Moissan used fluoride salts with electrolysis. Thus on June 28, 1886, he performed his experiment in front of the Académie des sciences to show his discovery of the new element fluorine.[11] While trying to find elemental fluorine through electrolysis of fluoride salts, many chemists perished including Paulin Louyet and Jérôme Nicklès.[12]
In 1886 Charles Martin Hall from America and Paul Héroult from France both filed for American patents for the electrolysis of aluminum, with Héroult submitting his in May, and Hall, in July.[13] Hall was able to get his patent by proving through letters to his brother and family evidence that his method was discovered before the French patent was submitted.[14] This became known as the Hall–Héroult process which benefited many industries because aluminum's price then dropped from four dollars to thirty cents per pound.[15]
In 1902 Polish engineer and inventor Stanisław Łaszczyński filed for and obtained Polish patent for the electrolysis of copper and zinc.[16][17][18]
Timeline
[edit]- 1785 – Martinus van Marum's electrostatic generator was used to reduce tin, zinc, and antimony from their salts using electrolysis.[19]
- 1789 - Adriaan Paets van Troostwijk and Jan Rudolph Deiman conducted early experiments on water electrolysis using an Electrostatic generator, producing small amounts of hydrogen and oxygen.[20][21]
- 1800 – William Nicholson and Anthony Carlisle (and also Johann Ritter), demonstrated the first sustained electrolysis of water into hydrogen and oxygen using a Voltaic pile.
- 1808 – Potassium (1807), sodium (1807), barium, calcium and magnesium were discovered by Humphry Davy using electrolysis.
- 1821 – Lithium was discovered by the English chemist William Thomas Brande, who obtained it by electrolysis of lithium oxide.
- 1834 – Michael Faraday published his two laws of electrolysis, provided a mathematical explanation for them, and introduced terminology such as electrode, electrolyte, anode, cathode, anion, and cation.
- 1875 – Paul Émile Lecoq de Boisbaudran discovered gallium using electrolysis.[22]
- 1886 – Fluorine was discovered by Henri Moissan using electrolysis.
- 1886 – Hall–Héroult process developed for making aluminium.
- 1890 – Castner–Kellner process developed for making sodium hydroxide.
- 1902 – Stanisław Łaszczyński obtained copper using electrolysis.[16][17][18]
- 1930 – Development of the modern chlor-alkali process (electrolysis of brine to produce chlorine and sodium hydroxide), which became an important industrial method.[23]
Overview
[edit]Electrolysis is the passing of a direct electric current through an electrolyte which is producing chemical reactions at the electrodes and decomposition of the materials.
The main components required to achieve electrolysis are an electrolyte, electrodes, and an external power source. A partition (e.g. an ion-exchange membrane or a salt bridge) is optional to keep the products from diffusing to the vicinity of the opposite electrode.
The electrolyte is a chemical substance which contains free ions and carries electric current (e.g. an ion-conducting polymer, solution, or an ionic liquid compound). If the ions are not mobile, as in most solid salts, then electrolysis cannot occur. A liquid electrolyte is produced by:
- Solvation or reaction of an ionic compound with a solvent (such as water) to produce mobile ions
- An ionic compound melted by heating
The electrodes are immersed separated by a distance such that a current flows between them through the electrolyte and are connected to the power source which completes the electrical circuit. A direct current supplied by the power source drives the reaction causing ions in the electrolyte to be attracted toward the respective oppositely charged electrode.
Electrodes of metal, graphite and semiconductor material are widely used. Choice of suitable electrode depends on chemical reactivity between the electrode and electrolyte and manufacturing cost. Historically, when non-reactive anodes were desired for electrolysis, graphite (called plumbago in Faraday's time) or platinum were chosen.[24] They were found to be some of the least reactive materials for anodes. Platinum erodes very slowly compared to other materials, and graphite crumbles and can produce carbon dioxide in aqueous solutions but otherwise does not participate in the reaction. Cathodes may be made of the same material, or they may be made from a more reactive one since anode wear is greater due to oxidation at the anode.
Process of electrolysis
[edit]The key process of electrolysis is the interchange of atoms and ions by the removal or addition of electrons due to the applied potential. The desired products of electrolysis are often in a different physical state from the electrolyte and can be removed by mechanical processes (e.g. by collecting gas above an electrode or precipitating a product out of the electrolyte).
The quantity of the products is proportional to the current, and when two or more electrolytic cells are connected in series to the same power source, the products produced in the cells are proportional to their equivalent weight. These are known as Faraday's laws of electrolysis.
Each electrode attracts ions that are of the opposite charge. Positively charged ions (cations) move towards the electron-providing (negative) cathode. Negatively charged ions (anions) move towards the electron-extracting (positive) anode. In this process electrons are effectively introduced at the cathode as a reactant and removed at the anode as a product. In chemistry, the loss of electrons is called oxidation, while electron gain is called reduction.
When neutral atoms or molecules, such as those on the surface of an electrode, gain or lose electrons they become ions and may dissolve in the electrolyte and react with other ions.
When ions gain or lose electrons and become neutral, they will form compounds that separate from the electrolyte. Positive metal ions like Cu2+ deposit onto the cathode in a layer. The terms for this are electroplating, electrowinning, and electrorefining.
When an ion gains or loses electrons without becoming neutral, its electronic charge is altered in the process.
For example, the electrolysis of brine produces hydrogen and chlorine gases which bubble from the electrolyte and are collected. The initial overall reaction is thus:[25]
- 2 NaCl + 2 H2O → 2 NaOH + H2 + Cl2
The reaction at the anode results in chlorine gas from chlorine ions:
- 2 Cl− → Cl2 + 2 e−
The reaction at the cathode results in hydrogen gas and hydroxide ions:
- 2 H2O + 2 e− → H2 + 2 OH−
Without a partition between the electrodes, the OH− ions produced at the cathode are free to diffuse throughout the electrolyte to the anode. As the electrolyte becomes more basic due to the production of OH−, less Cl2 emerges from the solution as it begins to react with the hydroxide producing hypochlorite (ClO−) at the anode:
- Cl2 + 2 NaOH → NaCl + NaClO + H2O
The more opportunity the Cl2 has to interact with NaOH in the solution, the less Cl2 emerges at the surface of the solution and the faster the production of hypochlorite progresses. This depends on factors such as solution temperature, the amount of time the Cl2 molecule is in contact with the solution, and concentration of NaOH.
Likewise, as hypochlorite increases in concentration, chlorates are produced from them:
- 3 NaClO → NaClO3 + 2 NaCl
Other reactions occur, such as the self-ionization of water and the decomposition of hypochlorite at the cathode, the rate of the latter depends on factors such as diffusion and the surface area of the cathode in contact with the electrolyte.[26]
Decomposition potential
[edit]Decomposition potential or decomposition voltage refers to the minimum voltage (difference in electrode potential) between anode and cathode of an electrolytic cell that is needed for electrolysis to occur.[27]
The voltage at which electrolysis is thermodynamically preferred is the difference of the electrode potentials as calculated using the Nernst equation. Applying additional voltage, referred to as overpotential, can increase the rate of reaction and is often needed above the thermodynamic value. It is especially necessary for electrolysis reactions involving gases, such as oxygen, hydrogen or chlorine.
Oxidation and reduction at the electrodes
[edit]Oxidation of ions or neutral molecules occurs at the anode. For example, it is possible to oxidize ferrous ions to ferric ions at the anode:
- Fe2+
(aq) → Fe3+
(aq) + e−
Reduction of ions or neutral molecules occurs at the cathode. It is possible to reduce ferricyanide ions to ferrocyanide ions at the cathode:
- Fe(CN)3-
6 + e− → Fe(CN)4-
6
Neutral molecules can also react at either of the electrodes. For example: p-benzoquinone can be reduced to hydroquinone at the cathode:
 + 2 e− + 2 H+ →
+ 2 e− + 2 H+ → 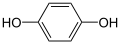
In the last example, H+ ions (hydrogen ions) also take part in the reaction and are provided by the acid in the solution, or by the solvent itself (water, methanol, etc.). Electrolysis reactions involving H+ ions are fairly common in acidic solutions. In aqueous alkaline solutions, reactions involving OH− (hydroxide ions) are common.
Sometimes the solvents themselves (usually water) are oxidized or reduced at the electrodes. It is even possible to have electrolysis involving gases, e.g. by using a gas diffusion electrode.
Energy changes during electrolysis
[edit]The amount of electrical energy that must be added equals the change in Gibbs free energy of the reaction plus the losses in the system. The losses can (in theory) be arbitrarily close to zero, so the maximum thermodynamic efficiency equals the enthalpy change divided by the free energy change of the reaction. In most cases, the electric input is larger than the enthalpy change of the reaction, so some energy is released in the form of heat. In some cases, for instance, in the electrolysis of steam into hydrogen and oxygen at high temperature, the opposite is true and heat energy is absorbed. This heat is absorbed from the surroundings, and the heating value of the produced hydrogen is higher than the electric input.
Variations
[edit]Pulsating current results in products different from DC. For example, pulsing increases the ratio of ozone to oxygen produced at the anode in the electrolysis of an aqueous acidic solution such as dilute sulphuric acid.[28] Electrolysis of ethanol with pulsed current evolves an aldehyde instead of primarily an acid.[29]
Related processes
[edit]Galvanic cells and batteries use spontaneous, energy-releasing redox reactions to generate an electrical potential that provides useful power. When a secondary battery is charged, its redox reaction is run in reverse and the system can be considered as an electrolytic cell.
Industrial uses
[edit]The chloralkali process is a large scale application of electrolysis. This technology supplies most of the chlorine and sodium hydroxide required by many industries. The cathode is a mixed metal oxide clad titanium anode (also called a dimensionally stable anode).[30][31]

Electrofluorination
[edit]Many organofluorine compounds are produced by electrofluorination.[32] One manifestation of this technology is the Simons process, which can be described as:
- R3C–H + HF → R3C–F + H2
In the course of a typical synthesis, this reaction occurs once for each C–H bond in the precursor. The cell potential is maintained near 5–6 V. The anode, the electrocatalyst, is nickel-plated.
Hydrodimerization of acrylonitrile
[edit]Acrylonitrile is converted to adiponitrile on an industrial scale via electrocatalysis.[33]

Electroplating and electrowinning processes
[edit]- Purifying copper from refined copper.
- Electrometallurgy of aluminium, lithium, sodium, potassium, magnesium, calcium.
Electroplating, where a thin film of metal is deposited over a substrate material. Electroplating is used in many industries for either functional or decorative purposes, as in-vehicle bodies and nickel coins.
Electrochemical machining (ECM)
[edit]In Electrochemical machining, an electrolytic cathode is used as a shaped tool for removing material by anodic oxidation from a workpiece. ECM is often used as a technique for deburring or for etching metal surfaces like tools or knives with a permanent mark or logo.
Other
[edit]- Production of sodium chlorate and potassium chlorate.
- Production of fuels such as hydrogen for spacecraft, nuclear submarines and vehicles.
- Rust removal and cleaning of old coins and other metallic objects.
Competing half-reactions in solution electrolysis
[edit]Using a cell containing inert platinum electrodes, electrolysis of aqueous solutions of some salts leads to the reduction of the cations (such as metal deposition with, for example, zinc salts) and oxidation of the anions (such as the evolution of bromine with bromides). However, with salts of some metals (such as sodium) hydrogen is evolved at the cathode, and for salts containing some anions (such as sulfate SO2−
4) oxygen is evolved at the anode. In both cases, this is due to water being reduced to form hydrogen or oxidized to form oxygen.
In principle, the voltage required to electrolyze a salt solution can be derived from the standard electrode potential for the reactions at the anode and cathode. The standard electrode potential is directly related to the Gibbs free energy, ΔG, for the reactions at each electrode and refers to an electrode with no current flowing. An extract from the table of standard electrode potentials is shown below.
Half-reaction E° (V) Ref. Na+ + e− ⇌ Na(s) −2.71 [34] Zn2+ + 2 e− ⇌ Zn(s) −0.7618 [35] 2 H+ + 2 e− ⇌ H2(g) ≡ 0 [35] Br2(aq) + 2 e− ⇌ 2 Br− +1.0873 [35] O2(g) + 4 H+ + 4 e− ⇌ 2 H2O +1.23 [34] Cl2(g) + 2 e− ⇌ 2 Cl− +1.36 [34] S
2O2−
8 + 2 e− ⇌ 2 SO2−
4+2.07 [34]
In terms of electrolysis, this table should be interpreted as follows:
- Moving down the table, E° becomes more positive, and species on the left are more likely to be reduced: for example, zinc ions are more likely to be reduced to zinc metal than sodium ions are to be reduced to sodium metal.
- Moving up the table, E° becomes more negative, and species on the right are more likely to be oxidized: for example, sodium metal is more likely to be oxidized to sodium ions than zinc metal is to be oxidized to zinc ions.
Using the Nernst equation the electrode potential can be calculated for a specific concentration of ions, temperature and the number of electrons involved. For pure water (pH 7):
- the electrode potential for the reduction producing hydrogen is −0.41 V,
- the electrode potential for the oxidation producing oxygen is +0.82 V.
Comparable figures calculated in a similar way, for 1 M zinc bromide, ZnBr2, are −0.76 V for the reduction to Zn metal and +1.10 V for the oxidation producing bromine. The conclusion from these figures is that hydrogen should be produced at the cathode and oxygen at the anode from the electrolysis of water—which is at variance with the experimental observation that zinc metal is deposited and bromine is produced.[36] The explanation is that these calculated potentials only indicate the thermodynamically preferred reaction. In practice, many other factors have to be taken into account such as the kinetics of some of the reaction steps involved. These factors together mean that a higher potential is required for the reduction and oxidation of water than predicted, and these are termed overpotentials. Experimentally it is known that overpotentials depend on the design of the cell and the nature of the electrodes.
For the electrolysis of a neutral (pH 7) sodium chloride solution, the reduction of sodium ion is thermodynamically very difficult and water is reduced evolving hydrogen leaving hydroxide ions in solution. At the anode the oxidation of chlorine is observed rather than the oxidation of water since the overpotential for the oxidation of chloride to chlorine is lower than the overpotential for the oxidation of water to oxygen. The hydroxide ions and dissolved chlorine gas react further to form hypochlorous acid. The aqueous solutions resulting from this process is called electrolyzed water and is used as a disinfectant and cleaning agent.
Research trends
[edit]Electrolysis of carbon dioxide
[edit]The electrochemical reduction or electrocatalytic conversion of CO2 can produce value-added chemicals such as methane, ethylene, ethanol, etc.[37][38][39] The electrolysis of carbon dioxide gives formate or carbon monoxide, but sometimes more elaborate organic compounds such as ethylene.[40] This technology is under research as a carbon-neutral route to organic compounds.[41][42]
Electrolysis of acidified water
[edit]Electrolysis of water produces hydrogen and oxygen in a ratio of 2 to 1 respectively.
- 2 H2O(l) → 2 H2(g) + O2(g) E° = +1.229 V
The energy efficiency of water electrolysis varies widely. The efficiency of an electrolyser is a measure of the enthalpy contained in the hydrogen (to undergo combustion with oxygen or some other later reaction), compared with the input electrical energy. Heat/enthalpy values for hydrogen are well published in science and engineering texts, as 144 MJ/kg (40 kWh/kg). Note that fuel cells (not electrolysers) cannot use this full amount of heat/enthalpy, which has led to some confusion when calculating efficiency values for both types of technology. In the reaction, some energy is lost as heat. Some reports quote efficiencies between 50% and 70% for alkaline electrolysers (50 kWh/kg);[43] however, higher practical efficiencies are available with the use of polymer electrolyte membrane electrolysis and catalytic technology, such as 95% efficiency.[44][45]
The National Renewable Energy Laboratory estimated in 2006 that 1 kg of hydrogen (roughly equivalent to 3 kg, or 4 liters, of petroleum in energy terms) could be produced by wind powered electrolysis for between US$5.55 in the near term and US$2.27 in the longer term.[46]
About 4% of hydrogen gas produced worldwide is generated by electrolysis, and normally used onsite. Hydrogen is used for the creation of ammonia for fertilizer via the Haber process, and converting heavy petroleum sources to lighter fractions via hydrocracking. Onsite electrolysis has been utilized to capture hydrogen for hydrogen fuel-cells in hydrogen vehicles.
Carbon/hydrocarbon assisted water electrolysis
[edit]Recently, to reduce the energy input, the utilization of carbon (coal), alcohols (hydrocarbon solution), and organic solution (glycerol, formic acid, ethylene glycol, etc.) with co-electrolysis of water has been proposed as a viable option.[47][48] The carbon/hydrocarbon assisted water electrolysis (so-called CAWE) process for hydrogen generation would perform this operation in a single electrochemical reactor. This system energy balance can be required only around 40% electric input with 60% coming from the chemical energy of carbon or hydrocarbon.[49] This process utilizes solid coal/carbon particles or powder as fuels dispersed in acid/alkaline electrolyte in the form of slurry and the carbon contained source co-assist in the electrolysis process as following theoretical overall reactions:[50]
- Carbon/Coal slurry (C + 2H2O) → CO2 + 2H2 E′ = 0.21 V (reversible voltage) / E′ = 0.46 V (thermo-neutral voltage)
or
- Carbon/Coal slurry (C + H2O) → CO + H2 E′ = 0.52 V (reversible voltage) / E′ = 0.91 V (thermo-neutral voltage)
Thus, this CAWE approach is that the actual cell overpotential can be significantly reduced to below 1.0 V as compared to 1.5 V for conventional water electrolysis.
Electrocrystallization
[edit]A specialized application of electrolysis involves the growth of conductive crystals on one of the electrodes from oxidized or reduced species that are generated in situ. The technique has been used to obtain single crystals of low-dimensional electrical conductors, such as charge-transfer salts and linear chain compounds.[51][52]
Electrolysis of Iron Ore
[edit]The current method of producing steel from iron ore is very carbon intensive, in part to the direct release of CO2 in the blast furnace. A study of steel making in Germany found that producing 1 ton of steel emitted 2.1 tons of CO2e with 22% of that being direct emissions from the blast furnace.[53] As of 2022, steel production contributes 7–9% of global emissions.[54] Electrolysis of iron can eliminate direct emissions and further reduce emissions if the electricity is created from green energy.
The small-scale electrolysis of iron has been successfully reported by dissolving it in molten oxide salts and using a platinum anode.[55] Oxygen anions form oxygen gas and electrons at the anode. Iron cations consume electrons and form iron metal at the cathode. This method was performed a temperature of 1550 °C which presents a significant challenge to maintaining the reaction. Particularly, anode corrosion is a concern at these temperatures.
Additionally, the low temperature reduction of iron oxide by dissolving it in alkaline water has been reported.[56] The temperature is much lower than traditional iron production at 114 °C. The low temperatures also tend to correlate with higher current efficiencies, with an efficiency of 95% being reported. While these methods are promising, they struggle to be cost competitive because of the large economies of scale keeping the price of blast furnace iron low.
Electrolysis of seawater
[edit]A 2020 study investigated direct electrolysis of seawater, alkaline electrolysis, proton-exchange membrane electrolysis, and solid oxide electrolysis.[57] Direct electrolysis of seawater follows known processes, forming an electrolysis cell in which the seawater acts as the electrolyte to allow for the reaction at the anode, 2 Cl−(aq) → Cl2(g) + 2e− and the reaction at the cathode, 2 H2O(l) + 2 e− → H2(g) + 2OH−(aq). The inclusion of magnesium and calcium ions in the seawater makes the production of alkali hydroxides possible that could form scales in the electrolyser cell, cutting down on lifespan and increasing the need for maintenance. The alkaline electrolysers operate with the following reactions at the anode, 2 OH−(aq) → 1/2 O2(g) + H2O(l) + 2 e−, and at the cathode, 2 H2O(l) + 2 e− → H2(g) + 2 OH−(aq), and use high base solutions as electrolytes, operating at 60–90 °C (140–194 °F) and need additional separators to ensure the gas phase hydrogen and oxygen remain separate. The electrolyte can easily get contaminated, but the alkaline electrolyser can operate under pressure to improve energy consumption. The electrodes can be made of inexpensive materials and there's no requirement for an expensive catalyst in the design.
Many alternatives to this simple electrolyzer described exist. Micro-electrolyzer designs are able to eliminate the separator requirement by designing the internal flow to separate the gases autonomously. See for example US 12116679B2 ("Device and method for large scale harvesting of solar energy through hydrogen production") where the operating pressure is increased to the point of Chlorine liquefaction so that sea water electrolyzer can proceed in a locally alkaline electrolytic fluid. Removing separators allows operating at very high temperatures. The structural design allows for operations at upto 700 bar thereby eliminating the need for Hydrogen compressors.
Proton-exchange membrane electrolysers operate with the reactions at the anode, H2O(l) → 1/2 O2(g) + 2 H+(aq) + 2 e− and cathode, 2 H+(aq) + 2 e− → H2(g), at temperatures of 60–80 °C (140–176 °F), using a solid polymer electrolyte and requiring higher costs of processing to allow the solid electrolyte to touch uniformly to the electrodes. Similar to the alkaline electrolyser, the proton exchange membrane electrolyser can operate at higher pressures, reducing the energy costs required to compress the hydrogen gas afterward, but the proton exchange membrane electrolyser also benefits from rapid response times to changes in power requirements or demands and not needing maintenance, at the cost of having a faster inherent degradation rate and being the most vulnerable to impurities in the water.
Solid oxide electrolysers run the reactions O−2(g) → 1/2 O2(g) + 2 e− at the anode and H2O(g) + 2 e− → H2(g) + O−2(g) at the cathode. The solid oxide electrolysers require high temperatures (700–1,000 °C (1,292–1,832 °F)) to operate, generating superheated steam. They suffer from degradation when turned off, making it a more inflexible hydrogen generation technology. In a selected series of multiple-criteria decision-analysis comparisons in which the highest priority was placed on economic operation costs followed equally by environmental and social criteria, it was found that the proton exchange membrane electrolyser offered the most suitable combination of values (e.g., investment cost, maintenance, and operation cost, resistance to impurities, specific energy for hydrogen production at sea, risk of environmental impact, etc.), followed by the alkaline electrolyser, with the alkaline electrolyser being the most economically feasible, but more hazardous in terms of safety and environmental concerns due to the need for basic electrolyte solutions as opposed to the solid polymers used in proton-exchange membranes. Due to the methods conducted in multiple-criteria decision analysis, non-objective weights are applied to the various factors, and so multiple methods of decision analysis were performed simultaneously to examine the electrolysers in a way that minimizes the effects of bias on the performance conclusions.
See also
[edit]- Alkaline water electrolysis
- Castner–Kellner process
- Combined cycle hydrogen power plant
- Electrolytic cell
- Electrochemical engineering
- Faraday's laws of electrolysis
- Faraday constant
- Faraday efficiency
- Galvanic corrosion
- Galvanoluminescence
- Gas cracker
- Hall–Héroult process
- High-pressure electrolysis
- Patterson Power Cell
- Thermochemical cycle
- Timeline of hydrogen technologies
- PEM electrolysis
References
[edit]- ^ Oesper, Ralph; Speter, Max (1937). "The Faraday-Whewell correspondence concerning electro-chemical terms". The Scientific Monthly. 45 (6): 535–546. Bibcode:1937SciMo..45..535O.
- ^ Fabbri, Emiliana; Schmidt, Thomas J. (5 October 2018). "Oxygen Evolution Reaction—The Enigma in Water Electrolysis". ACS Catalysis. 8 (10): 9765–9774. doi:10.1021/acscatal.8b02712.
- ^ Ashworth, William (20 March 2015). "Martinus van Marum - Scientist of the Day". Linda Hall Library.
- ^ Ihde, Aaron J. (1964). The Development of Modern Chemistry. Harper & Row. pp. 125–127.
- ^ a b "The History of Electrochemistry: From Volta to Edison". ECS. Retrieved 11 October 2019.
- ^ Thorpe, Thomas (1896). Humphry Davy, Poet and Philosopher. New York: Macmillan & Co., Limited.
- ^ "Lithium - periodicstats.com". www.periodicstats.com. Archived from the original on 15 April 2021.
- ^ Helmenstine, Todd (11 January 2018). "Today in Science History - January 12 - Johan August Arfwedson and Lithium". Science Notes and Projects.
- ^ Ede, Andrew (2017). A History of Science in Society (3rd ed.). Toronto, Canada: University of Toronto Press. p. 251. ISBN 978-1-4426-3499-2.
- ^ Marshall, James and Virginia. "Rediscovery of the Elements – Gallium" (PDF). University of North Texas. Archived (PDF) from the original on 9 October 2022.
- ^ Wisniak, Jaime (26 August 2018). "Henri Moissan: The discoverer of fluorine". Educación Química. 13 (4): 267. doi:10.22201/fq.18708404e.2002.4.66285. ISSN 1870-8404. S2CID 92299077.
- ^ Marshall, James and Virginia. "Rediscovery of the Elements- Fluorine and Henri Moissan" (PDF). University of North Texas. Archived (PDF) from the original on 9 October 2022.
- ^ Beck, Theodore R. (21 August 2015). "ECS Classics: Hall and Héroult and the Discovery of Aluminum Electrolysis". ECS. Interface.
- ^ Hall Process Production and Commercialization of Aluminum - National Historic Chemical Landmark. Oberlin, Ohio: American Chemical Society. 1997.
- ^ "Paul Héroult and Charles M. Hall". Science History Institute. 1 June 2016.
- ^ a b "Bohaterski naukowiec upamiętniony na kieleckim deptaku" (in Polish). 18 September 2019. Retrieved 3 June 2024.
- ^ a b "De Verbno Łaszczyńscy » Stanisław Ignacy Łaszczyński" (in Polish). Retrieved 3 June 2024.
- ^ a b "Wyborcza.pl". wyborcza.pl. Retrieved 3 June 2024.
- ^ The Supplement (1803 edition) to Encyclopædia Britannica 3rd edition (1797), volume 1, page 225, "Mister Van Marum, by means of his great electrical machine, decomposed the calces of tin, zinc, and antimony, and resolved them into their respective metals and oxygen" and gives as a reference Journal de Physiques, 1785.
- ^ van Troostwijk, A. Paets; Deiman, J.R. (1789). "Sur une manière de décomposer l'eau en air inflammable et en air vital". Journal de Physique. 35.
- ^ de Levie, R (October 1999). "The electrolysis of water". Journal of Electroanalytical Chemistry. 476 (1): 92–93. doi:10.1016/s0022-0728(99)00365-4. ISSN 1572-6657.
- ^ Lecoq de Boisbaudran P-E (1875). "On certain properties of gallium". The Chemical News and Journal of Industrial Science. 32: 294.
- ^ Electrochemical Society. (n.d.). Metals & Chemicals. Retrieved February 2, 2025, fromElectrochemical Society. (n.d.). Metals & Chemicals. Retrieved https://www1.eere.energy.gov/manufacturing/resources/chemicals/pdfs/profile_chap6.pdf
- ^ Faraday, Michael (1849). Experimental Researches In Electricity. Vol. 1. London: The University of London.
- ^ Tilley, R.J.D. (2004). Understanding solids: the science of materials. John Wiley and Sons. pp. 281–. Bibcode:2004usts.book.....T. ISBN 978-0-470-85276-7. Retrieved 22 October 2011.
- ^ Thompson, M. de Kay (1911). Applied Electrochemistry. The MacMillan Company. pp. 84-88.
- ^ Blum, W.; Vinal, G. W. (1934). "The Definition of Polarization, Overvoltage and Decomposition Potential". Transactions of the Electrochemical Society. 66: 359. doi:10.1149/1.3498105.
- ^ Kremann, R. (1903). "Über den Einfluss der Natur des Elektrolyten und des Elektrodenmaterials auf die Ozonbildung". Zeitschrift für anorganische Chemie. 36. Metzger & Wittig: 403–. doi:10.1002/zaac.19030360152. Retrieved 12 September 2019.
- ^ Hale, Arthur J. (1919). The Manufacture of Chemicals by Electrolysis. D. Van Nostrand Co. pp. 16, 73. Retrieved 12 September 2019.
- ^ Over, Herbert (2012). "Surface Chemistry of Ruthenium Dioxide in Heterogeneous Catalysis and Electrocatalysis: From Fundamental to Applied Research". Chemical Reviews. 112 (6): 3356–3426. doi:10.1021/cr200247n. PMID 22423981.
- ^ Landolt, D.; Ibl, N. (1972). "Anodic Chlorate Formation on Platinized Titanium". Journal of Applied Electrochemistry. 2 (3). Chapman and Hall Ltd.: 201–210. doi:10.1007/BF02354977. S2CID 95515683.
- ^ Siegemund, Günter; Schwertfeger, Werner; Feiring, Andrew; Smart, Bruce; Behr, Fred; Vogel, Herward; McKusick, Blaine (2000). "Fluorine Compounds, Organic". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a11_349. ISBN 3527306730.
- ^ Kotrel, Stefan; BrUninger, Sigmar (2008). "Industrial Electrocatalysis". Handbook of Heterogeneous Catalysis. doi:10.1002/9783527610044.hetcat0103. ISBN 978-3-527-31241-2.
- ^ a b c d Atkins, Peter (1997). Physical Chemistry (6 ed.). New York: W.H. Freeman and Company.
- ^ a b c Vanýsek, Petr (2007). "Electrochemical Series". Handbook of Chemistry and Physics (88 ed.). Chemical Rubber Company. Archived from the original (PDF) on 24 July 2017.
- ^ Hall, Norris F. (1952). "A Textbook of Quantitative Inorganic Analysis (Vogel, Arthur I.)". Journal of Chemical Education. 29 (6): 319. Bibcode:1952JChEd..29..319H. doi:10.1021/ed029p319.1.
- ^ Lee, Seunghwa; Ju, Hyungkuk; Machunda, Revocatus; Uhm, Sunghyun; Lee, Jae Kwang; Lee, Hye Jin; Lee, Jaeyoung (2015). "Sustainable production of formic acid by electrolytic reduction of gaseous carbon dioxide". J. Mater. Chem. A. 3 (6): 3029. doi:10.1039/C4TA03893B. S2CID 98110035.
- ^ Whipple, Devin T.; Kenis, Paul J.A. (2010). "Prospects of CO2 Utilization via Direct Heterogeneous Electrochemical Reduction". The Journal of Physical Chemistry Letters. 1 (24): 3451. doi:10.1021/jz1012627. S2CID 101946630.
- ^ Machunda, Revocatus L.; Ju, Hyungkuk; Lee, Jaeyoung (2011). "Electrocatalytic reduction of CO2 gas at Sn based gas diffusion electrode". Current Applied Physics. 11 (4): 986. Bibcode:2011CAP....11..986M. doi:10.1016/j.cap.2011.01.003.
- ^ Hori, Y (2008). "Electrochemical CO2 Reduction on Metal Electrodes". In C.G. Vayeanas, R. White and M.E. Gamboa-Aldeco (ed.). Modern Aspects of Electrochemistry. Vol. 42 (42 ed.). New York: Springer. pp. 141–153. doi:10.1007/978-0-387-49489-0_3. ISBN 978-0-387-49488-3..
- ^ Appel, Aaron M.; Bercaw, John E.; Bocarsly, Andrew B.; Dobbek, Holger; Dubois, Daniel L.; Dupuis, Michel; Ferry, James G.; Fujita, Etsuko; Hille, Russ; Kenis, Paul J.A.; Kerfeld, Cheryl A.; Morris, Robert H.; Peden, Charles H.F.; Portis, Archie R.; Ragsdale, Stephen W.; Rauchfuss, Thomas B.; Reek, Joost N.H.; Seefeldt, Lance C.; Thauer, Rudolf K.; Waldrop, Grover L. (2013). "Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation". Chemical Reviews. 113 (8): 6621–6658. doi:10.1021/cr300463y. PMC 3895110. PMID 23767781.
- ^ Qiao, Jinli; Liu, Yuyu; Hong, Feng; Zhang, Jiujun (2014). "A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels". Chem. Soc. Rev. 43 (2): 631–675. doi:10.1039/C3CS60323G. PMID 24186433.
- ^ Martin, Polly (5 July 2023). "Green hydrogen | Which type of electrolyser should you use? Alkaline, PEM, solid oxide or the latest tech?". rechargenews.com.
- ^ Carmo, M; Fritz D; Mergel J; Stolten D (2013). "A comprehensive review on PEM water electrolysis". Journal of Hydrogen Energy. 38 (12): 4901. Bibcode:2013IJHE...38.4901C. doi:10.1016/j.ijhydene.2013.01.151.
- ^ Zittel, Werner; Wurster, Reinhold (8 July 1996). "Chapter 3: Production of Hydrogen. Part 4: Production from electricity by means of electrolysis". HyWeb: Knowledge – Hydrogen in the Energy Sector. Ludwig-Bölkow-Systemtechnik GmbH. Archived from the original on 7 February 2007.
- ^ Levene, J.; Kroposki, B.; Sverdrup, G. (March 2006). "Wind Energy and Production of Hydrogen and Electricity – Opportunities for Renewable Hydrogen – Preprint" (PDF). National Renewable Energy Laboratory. Archived (PDF) from the original on 9 October 2022. Retrieved 20 October 2008.
- ^ Ju, Hyungkuk; Badwal, Sukhvinder; Giddey, Sarbjit (2018). "A comprehensive review of carbon and hydrocarbon assisted water electrolysis for hydrogen production". Applied Energy. 231: 502–533. Bibcode:2018ApEn..231..502J. doi:10.1016/j.apenergy.2018.09.125. S2CID 117669840.
- ^ Ju, Hyungkuk; Giddey, Sarbjit; Badwal, Sukhvinder P.S.; Mulder, Roger J. (2016). "Electro-catalytic conversion of ethanol in solid electrolyte cells for distributed hydrogen generation". Electrochimica Acta. 212: 744–757. doi:10.1016/j.electacta.2016.07.062.
- ^ Giddey, S.; Kulkarni, A.; Badwal, S.P.S. (2015). "Low emission hydrogen generation through carbon assisted electrolysis". International Journal of Hydrogen Energy. 40 (1): 70–74. Bibcode:2015IJHE...40...70G. doi:10.1016/j.ijhydene.2014.11.033.
- ^ Ju, Hyungkuk; Giddey, Sarbjit; Badwal, Sukhvinder P.S. (2018). "Role of iron species as mediator in a PEM based carbon-water co-electrolysis for cost-effective hydrogen production". International Journal of Hydrogen Energy. 43 (19): 9144–9152. Bibcode:2018IJHE...43.9144J. doi:10.1016/j.ijhydene.2018.03.195.
- ^ Bechgaard, K.; Carneiro, K.; Rasmussen, F. B.; Olsen, M.; Rindorf, G.; Jacobsen, C.S.; Pedersen, H.J.; Scott, J.C. (1981). "Superconductivity in an organic solid. Synthesis, structure, and conductivity of bis(tetramethyltetraselenafulvalenium) perchlorate, (TMTSF)2ClO4". Journal of the American Chemical Society. 103 (9): 2440. Bibcode:1981JAChS.103.2440B. doi:10.1021/ja00399a065.
- ^ Williams, Jack M. (2007). "Highly Conducting and Superconducting Synthetic Metals". Inorganic Syntheses. Vol. 26. pp. 386–394. doi:10.1002/9780470132579.ch70. ISBN 978-0-470-13257-9.
- ^ Backes, Jana Gerta; Suer, Julian; Pauliks, Nils; Neugebauer, Sabrina; Traverso, Marzia (19 March 2021). "Life Cycle Assessment of an Integrated Steel Mill Using Primary Manufacturing Data: Actual Environmental Profile". Sustainability. 13 (6): 3443. Bibcode:2021Sust...13.3443B. doi:10.3390/su13063443. ISSN 2071-1050.
- ^ Lopes, Daniela V.; Quina, Margarida J.; Frade, Jorge R.; Kovalevsky, Andrei V. (2022). "Prospects and challenges of the electrochemical reduction of iron oxides in alkaline media for steel production". Frontiers in Materials. 9. Bibcode:2022FrMat...910156L. doi:10.3389/fmats.2022.1010156. ISSN 2296-8016.
- ^ Wiencke, Jan; Lavelaine, Hervé; Panteix, Pierre-Jean; Petitjean, Carine; Rapin, Christophe (1 January 2018). "Electrolysis of iron in a molten oxide electrolyte". Journal of Applied Electrochemistry. 48 (1): 115–126. doi:10.1007/s10800-017-1143-5. ISSN 1572-8838. S2CID 102871146.
- ^ Yuan, Boyan; Haarberg, Geir Martin (20 March 2009). "Electrodeposition of Iron in Aqueous Alkaline Solution: An Alternative to Carbothermic Reduction". ECS Transactions. 16 (36): 31. Bibcode:2009ECSTr..16J..31Y. doi:10.1149/1.3114006. ISSN 1938-5862. S2CID 96771590.
- ^ d'Amore-Domenech, Rafael; Santiago, Óscar; Leo, Teresa J. (2020). "Multicriteria analysis of seawater electrolysis technologies for green hydrogen production at sea". Renewable and Sustainable Energy Reviews. 133 110166. Bibcode:2020RSERv.13310166D. doi:10.1016/j.rser.2020.110166. S2CID 224843343.
| International | |
|---|---|
| National | |
| Other | |